
Cardiac amyloidosis is a systemic disease which affects the heart tissue. You may also hear it referred to as “stiff heart syndrome”.While it is relatively rare, if left unrecognized or untreated, it can progress to causing arrhythmias or congestive heart failure.Here is what cardiologists should know:
What is cardiac amyloidosis?
Cardiac amyloidosis is a disorder of the heart that is caused by an abnormal protein build up within the heart tissue. This protein build up or plaque makes it difficult for the heart to function properly. Amyloidosis itself is a group of diseases where the proteins (amyloids) clump and build up in bodily organs. Over time, the proteins replace normal tissue, meaning the organ involved fails. Amyloidosis can affect the heart, kidneys, liver, spleen, nervous system, stomach or intestines.
Prevalence of cardiac amyloidosis
The exact numbers around the prevalence of cardiac amyloidosis are difficult to quantify due to changing diagnostic criteria. In the United States, around 2000 – 2500 cases of AL (light chain amyloidosis) are diagnosed each year. In the U.K. amyloidosis is listed as the cause of death in around 1 in 1000 people.Overall, cardiac amyloidosis is rare in patients under the age of 40. The following table shows common amyloid subtypes and clinical characteristics:
AL cardiac amyloidosis impacts roughly 10 out of every 100 individuals (estimates range between 8-12:1,000) with the majority of diagnosis being made between the ages of 55 to 60. ATTR cardiac amyloidosis generally affects patients older than 60 years of age and 25% of the population, age 80+, have their myocardium infiltrated by ATTR amyloid deposits.As more research is published, the data identifies cardiac amyloidosis as predominantly under-diagnosed, especially in the elderly population with heart failure. It is important to note that the frequency of diagnosing cardiac amyloidosis is on the rise as more awareness and data is available to physicians.
Causes of cardiac amyloidosis
There are dozens of causes of cardiac amyloidosis but the majority of incidences are caused in one of two ways:
Primary Amyloidosis
One common form of cardiac amyloidosis is diagnosed as light chain amyloidosis (AL) or primary amyloidosis. AL originates in the plasma cells which produce heavy chains and light chains. When these plasma cells become clonal, an overabundance of light chains may occur. Once this happens, there are three possible outcomes: 1) The light chain is excreted in the urine and is harmless 2) The plasma cell clone grows rapidly and takes over a portion of the bone marrow; potentially leading to myeloma 3) The light chain accumulates into beta-pleated sheets, circulate within the bloodstream and are deposited in one or more tissues. When the latter occurs within the cardiac tissue, it is referred to as cardiac amyloidosis.
Transthyretin amyloidosis
Transthyretin amyloidosis (ATTR) is caused by excess deposits of the normally occurring protein produced by the liver called transthyretin. These protein deposits may occur in soft tissue and vasculature, resulting in different diagnosis, depending on the area that is impacted. When in the soft tissue, an individual may develop a condition such as carpal tunnel syndrome. When these deposits occur within the cardiac tissue, it is referred to as transthyretin amyloidosis. There are two main ways in which a patient is categorized as having ATTR. The first, referred to as wild-type ATTR, occurs over a period of time where the protein build up eventually interferes with the function of the cardiac tissue. The other form of ATTR, mutant ATTR, occurs at an accelerated rate, much faster than wild-type and is due to a pathologic mutation of the transthyretin gene.
Identifying cardiac amyloidosis
Cardiac amyloidosis may or may not present with any symptoms in the patient. If symptoms are present, these might include:
- Fatigue and reduced exercise ability
- Heart palpitations
- Shortness of breath with activity
- Swelling in different parts of the body.
Definitive diagnosis of amyloidosis requires biopsy. Depending on the facility, physician preference and technology available, the physician may biopsy an area within the fat pad of the abdomen, bone marrow or the cardiac tissue directly. While the fat pad may be the easiest to access, the cardiac tissue biopsies provide a much higher sensitivity and specificity. Due to the invasiveness of this, physicians may opt for other, less invasive procedures to diagnose a patient.As with many disease states, there are laboratory tests that are available and helpful in determining which patients may be at high risk for developing the condition. The excess proteins involved in cardiac amyloidosis can be detected within the blood or urine. Other specialized blood tests are often monitored with patients who have cardiac amyloidosis; N-terminal pro-brain natriuretic peptide and troponin. These specialized tests provide information to physicians regarding how the heart is functioning and are often utilized in conjunction with therapy or treatment.While the biopsies and lab tests may help to identify the presence of cardiac amyloidosis and are required for a diagnosis, the best way to gauge the extent and impact of the disease is to visualize the cardiac tissue using common radiology exams. Many times, physicians will benefit from the information gathered during some or all of the following radiologic exams: echocardiography, cardiac magnetic resonance imaging (MRI) and radionuclide imaging. Currently, a “Gold Standard” imaging test has not been identified, however, radionuclide imaging for this disease state has been increasingly helpful.
Echocardiography
The information from this examination may include and are not limited to:
- Arterial septal wall thickness
- Left ventricle (LV) wall thickness
- LV end diastolic volume
- Left ventricle ejection fraction (LVEF)
- Transmitral flow
- Atrial and longitudinal strains.
MRI
The information from this examination may include and are not limited to:
- Diffuse decrease in the T1 and T2 signal intensity of the myocardium
- Left ventricular or atrial wall thickening
- Ejection fraction
- Diastolic filling
- Effusions – pericardial and pleural.
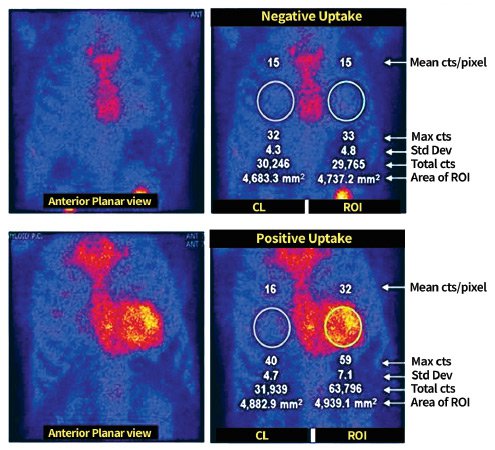
Radionuclide imaging (nuclear medicine)
An advantage unique to this procedure, utilizing 99Tc-PYP, is its ability to specifically identify ATTR cardiac amyloidosis non-invasively. The information from this examination may include and are not limited to:
- Accumulation and presence of ATTR proteins
- Differentiation between ATTR and AL cardiac amyloidosis
How is cardiac amyloidosis treated?
Treatment of AL cardiac amyloidosis is usually a multifaceted approach. To treat the root cause of the excess light chains, systemic chemotherapy is the general approach. Addressing the cardiac specific damage, physicians often involve volume management in the form of diuretics and salt restrictions and arrhythmia management.The FDA has recently approved medications for the treatment of ATTR cardiac amyloidosis and there are other medications that are being studied and are in late-phase clinical trials. In addition to medical intervention, one of the other common solutions to providing symptomatic relief is the use of a pacemaker, similar to AL cardiac amyloidosis.
Prognosis
The current general prognosis when there is cardiac involvement of amyloidosis is poor. The median survival rate without treatment is 13 months. Only around 5% of patients with AR survive beyond 10 years. Hopefully, with new medication being available for treatment, these statistics will improve.Complications as a result of cardiac amyloidosis include:
- Atrial fibrillation
- Congestive heart failure
- Embolism and stroke
- Ventricular arrhythmias
- Heart block requiring pacemaker implantation
- Pericardial tamponade
- Death
Final thoughts
Cardiac amyloidosis is a serious disease, most usually impacting patients over the age of 55. It is important for cardiologists to know the symptoms and diagnosis as it can lead to grave consequences for the patient.The current prognosis is poor, although there is some optimism with new treatments being trialed. The hope is that we will have more effective treatments in the future.
